Hypercholestérolémie familiale : la forme génétique à dépister
Une personne sur 250 porte une mutation qui fait exploser le LDL dès l'enfance. Critères DLCN, dépistage en cascade, statines précoces, inhibiteurs PCSK9 : la prise en charge 2026.

Souvent ignorée, l'HF reste sous-diagnostiquée : moins de 15 % des porteurs en France sont identifiés (NSFA, 2022). Pourtant, le diagnostic change radicalement la trajectoire de vie d'un patient et de sa famille, car il justifie une prévention intensive dès l'enfance.
Génétique et mécanismes
L'HF est due à une mutation d'un gène intervenant dans la captation du LDL par le foie :
- LDLR (gène du récepteur LDL) : 85 à 90 % des cas.
- APOB : 5 à 10 % — défaut d'ancrage du LDL au récepteur.
- PCSK9 gain de fonction : 1 à 3 % — dégradation accrue du récepteur.
- LDLRAP1 (forme récessive rare).
Forme hétérozygote (un seul allèle muté) : 1/250, LDL 1,9 à 4 g/L. Forme homozygote (les deux allèles) : 1/300 000, LDL 6 à 12 g/L, événement cardiovasculaire possible avant 20 ans.
Quand évoquer une HF ?
- LDL > 1,90 g/L chez l'adulte, > 1,50 g/L chez l'enfant, non expliqué par une cause secondaire.
- Antécédent familial de maladie coronaire précoce (< 55 ans homme, < 65 ans femme).
- Xanthomes tendineux (tendon d'Achille, extenseurs des doigts) — quasi-pathognomoniques.
- Arc cornéen avant 45 ans, xanthélasmas.
- LDL > 2,40 g/L = forte suspicion (probabilité > 80 %).
Scores diagnostiques validés
Deux outils de référence en Europe :
- DLCN (Dutch Lipid Clinic Network) : combine antécédents personnels, familiaux, examen clinique, LDL, génotype. Score ≥ 8 = HF certaine, 6-7 = probable, 3-5 = possible.
- Simon Broome : critères cliniques + familiaux + LDL + xanthomes + analyse génétique.
Le diagnostic moléculaire confirme dans 70 à 90 % des cas cliniquement certains (NGS d'un panel de 4 gènes, remboursé dans les centres de génétique agréés).
Dépistage en cascade — le levier décisif
Dès qu'un cas index est identifié, les apparentés du 1er degré (parents, enfants, fratrie) doivent être dépistés. Chaque apparenté a 50 % de risque d'être porteur. Le programme hollandais (Umans-Eckenhausen, Lancet 2001) a démontré qu'il permet de diagnostiquer 2 à 3 apparentés par cas index. En France, les réseaux HF (RHFCA, RegHF) coordonnent ce dépistage depuis 2015.
- 1ère étape : bilan lipidique + examen clinique chez tous les apparentés ≥ 5 ans.
- 2ème étape : test génétique ciblé sur la mutation familiale (coût divisé par 10).
Objectifs thérapeutiques
Selon l'EAS (2019) et la HAS (2017) :
- Adulte sans autre facteur : LDL < 1,0 g/L et baisse ≥ 50 %.
- Adulte avec facteur de risque majeur ou maladie CV : LDL < 0,55 g/L.
- Enfant ≥ 10 ans : LDL < 1,35 g/L, progressivement.
Stratégie médicamenteuse
La séquence habituelle :
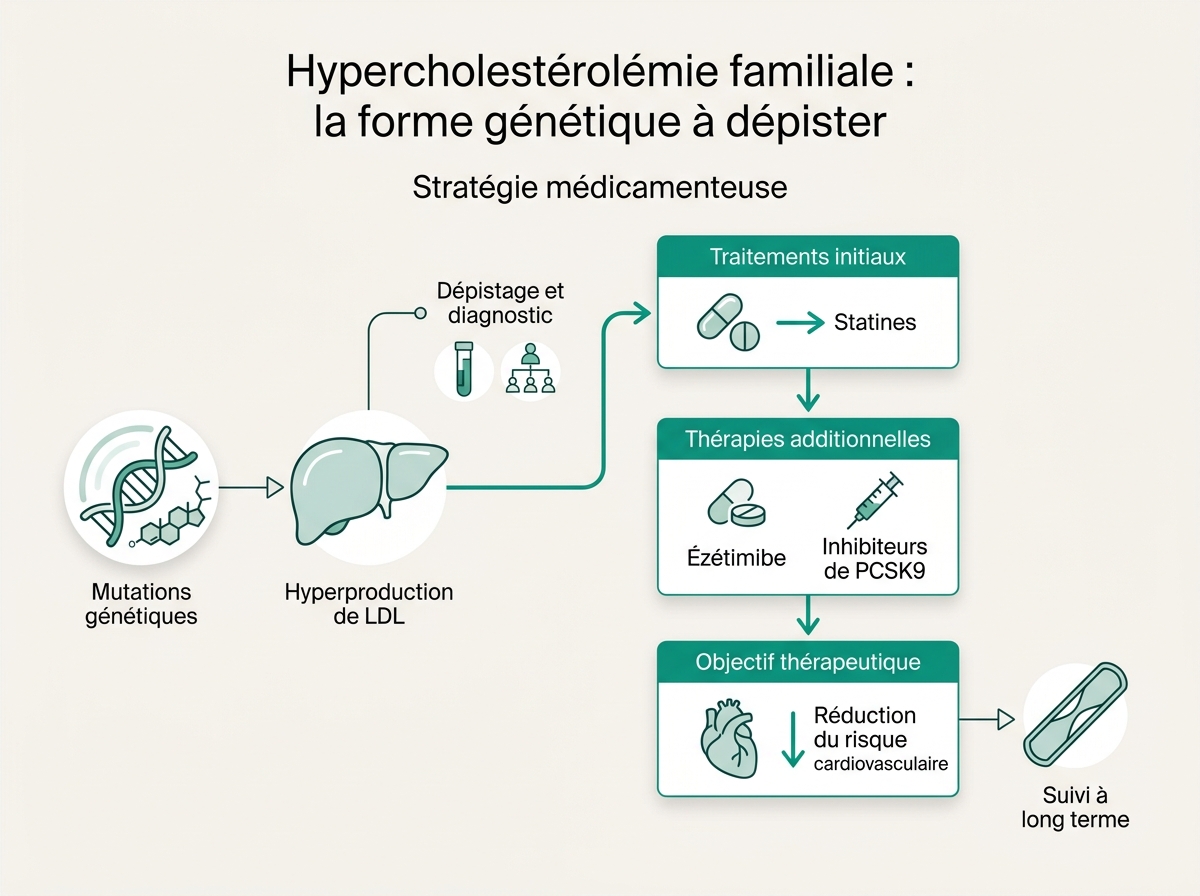
- Statine haute intensité d'emblée (atorvastatine 40-80 mg, rosuvastatine 20-40 mg) → baisse 50 %.
- Ézétimibe 10 mg en add-on → baisse complémentaire 15-20 %.
- Inhibiteur PCSK9 (évolocumab, alirocumab) s.c. toutes les 2 à 4 semaines → baisse 50-60 % supplémentaire. Remboursé en France chez l'HF hétérozygote à haut risque.
- Inclisiran (siRNA anti-PCSK9) : 2 injections par an après la dose de charge. AMM européenne 2020, remboursement HAS accordé en 2023 pour l'HF hétérozygote non contrôlée.
- Lomitapide : réservé à l'HF homozygote, centres experts.
- LDL-aphérèse : hémodialyse lipidique toutes les 1-2 semaines, indiquée dans l'HF homozygote ou hétérozygote sévère résistante.
Mesures hygiéno-diététiques
Indispensables en complément mais insuffisantes seules : elles baissent le LDL de 10 à 15 %, loin des 50 % attendus. Régime type Portfolio Diet, activité physique 150 min/semaine, sevrage tabagique strict, gestion tension et glycémie.
Suivi
- Bilan lipidique à 8 semaines après toute modification, puis tous les 6 à 12 mois.
- Bilan d'athérosclérose subclinique : IMT carotidien, score calcique coronarien à partir de 30-40 ans.
- Consultation de génétique médicale pour les projets de parentalité.
- Adhésion à un centre HF labellisé.
HF chez l'enfant
Dépister entre 5 et 10 ans si ATCD familial d'HF certaine. Statines autorisées à partir de 8-10 ans selon la molécule (pravastatine dès 8 ans, rosuvastatine dès 6 ans dans l'HF sévère). Le suivi pédiatrique spécialisé est recommandé par la NSFA.
Questions fréquentes
L'HF peut-elle être guérie ?
Non, c'est une mutation permanente. Mais le traitement médicamenteux à vie permet d'atteindre des LDL normaux et de ramener le risque cardiovasculaire à celui de la population générale. Les thérapies géniques (essai VERVE 2024) sont en développement.
Les enfants d'un parent HF doivent-ils être dépistés ?
Oui, dès 5 à 10 ans. Un bilan lipidique simple suffit en première intention ; le test génétique cible ensuite la mutation familiale connue. Cette démarche, appelée dépistage en cascade, évite des drames précoces et est soutenue par les réseaux HF français.
Peut-on avoir un LDL normal et être porteur d'HF ?
Exceptionnellement, les formes très atténuées existent (< 5 % des cas). Le test génétique reste le gold-standard. Sous traitement, le LDL peut redescendre à la normale — le diagnostic d'HF persiste.
L'inclisiran remplace-t-il les statines ?
Non, il s'ajoute. L'indication HAS prévoit l'inclisiran chez l'HF hétérozygote ne atteignant pas la cible malgré statine + ézétimibe à dose maximale tolérée. Deux injections par an facilitent grandement l'observance.
Que risque-t-on si l'HF n'est pas traitée ?
Sans traitement, la moitié des hommes HF font un infarctus avant 50 ans, et la moitié des femmes avant 60 ans (registre NorthStar, 2016). Les formes homozygotes peuvent présenter un infarctus dès l'adolescence. Le traitement précoce transforme ce pronostic.
Aller plus loin
- Cholestérol LDL et HDL : le guide complet1 — Pillar de référence.
- Bilan lipidique : interpréter ses résultats2 — Lire ses valeurs familiales.
- Cholestérol chez l'enfant et l'adolescent3 — Dépistage pédiatrique HF.
- Ézétimibe et inhibiteurs PCSK94 — Traitements de 2e ligne.
- Lipoprotéine(a) : le cholestérol génétique méconnu5 — Autre anomalie génétique à dépister.
Sources et références
- HAS — Prise en charge de l'hypercholestérolémie (2017)6
- NSFA — Société Française d'Athérosclérose : recommandations HF7
Recommandations NSFA sur le dépistage familial en cascade et la prise en charge de l'hypercholestérolémie familiale. - INSERM — Hypercholestérolémie familiale8
- ESC/EAS — Guidelines for the management of dyslipidaemias (2019)9
- Ameli — Prise en charge du cholestérol en cas d'HF10
Réponses aux questions les plus courantes
Articles Connexes
Découvrez d'autres articles sur ce sujet

Ramadan et diabète : adapter traitement, alimentation et risque hypoglycémique
Ramadan + diabète : ~ 70 % des diabétiques musulmans jeûnent. Stratification IDF-DAR 2021 (très élevé / élevé / modéré / faible). Adaptation antidiabétiques, surveillance glycémie capillaire. Sources HAS, IDF-DAR, EPIDIAR.

Prévenir la récidive de la lombalgie : 10 conseils evidence-based
Récidive lombalgique à 1 an : 60 %. Méta-analyse Steffens (JAMA 2016) : exercices + éducation réduisent la récidive de 25-45 %. STarT Back +33 % d'efficacité. Sources HAS, JAMA, Lancet, INSERM.

Matelas, oreiller et position de sommeil : impact réel sur le mal de dos
Matelas, oreiller, position de sommeil et lombalgie : ce que disent vraiment les méta-analyses. Fermeté médium > ferme. Position latérale > dorsale > ventrale. Sources HAS, Radwan 2015, Jacobson 2010, Bergholdt 2008.
Sourcé auprès d'autorités indépendantes
Cet article a été rédigé par Bilal YIKILMAZ, rédacteur en chef de cestlasante.com. Il n'est pas médecin : chaque recommandation ci-dessus s'appuie sur des sources médicales indépendantes, explicitement citées.
Autorités citées : HAS, INSERM, OMS.
Dernière révision éditoriale : .